

Abstract
Background:Improved survival with gemtuzumab ozogamicin (GO) in some people with acute myeloid leukemia has validated CD33 as immunotherapeutic target and sparked interested in developing new, highly potent CD33-directed therapeutics. As a limitation of this treatment strategy, CD33 expression on maturing and mature myeloid cells causes significant on-target, off-leukemia effects. Toxicity of CD33-targeted immunotherapy should be minimal in the presence of normal hematopoietic stem and progenitor cells (HSPCs) engineered to lack CD33 variants recognized by therapeutic antibodies. Indeed, very recent studies have shown that CRISPR/Cas9 with a single guide RNA (gRNA) designed to target the CD33coding region reduces display of CD33 and protects engineered cells from CD33 CAR T-cells. However, this approach resulted in low levels of CD33disruption in vivoand off-target activity. We therefore developed an approach in which the Cas9 protein is complexed with two synthetic gRNAs for precise excision of the intervening sequence (i.e. more controlled genome editing than what can be accomplished with a single gRNA). We directed these two gRNAs to intronic sequences for precise excision of exon 2, which encodes the V-set domain of CD33 that is recognized by all current CD33 therapeutics including GO. This approach eliminates exonic indels and protects engineered cells from CD33-targeted immunotherapy while maintaining expression of an exon 2-free variant of CD33 (CD33∆E2), which has previously been identified as natural isoform in human HSPCs.
Methods: We used human myeloid ML-1 cells and human fetal liver CD34+ HSPCs for CRISPR/Cas9 editing, which was carried out by electroporation of purified Cas9 protein complexed with synthetic gRNAs. In vitro cytotoxicity assays were performed to test sensitivity to GO and the CD33/CD3 bispecific antibody AMG 330.For in vivoassessment of engineered HSPCs, NSG neonate mice were infused with 6.0x105human CD34+ cells, and peripheral blood analyzed biweekly for 14 weeks.
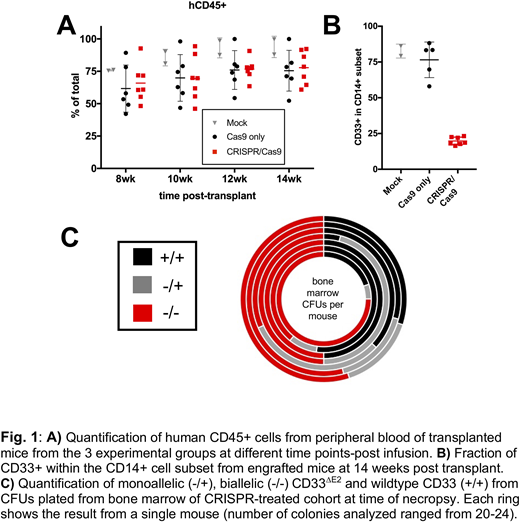
Results:Delivery of Cas9/sgRNA ribonucleoproteins (RNPs) in ML-1 cells resulted in exon 2 deletion and time-dependent reduction in cell surface display of full-length CD33 (CD33FL). Pure populations of CD33∆E2cells were generated via single-cell cloning. These sublines lacked surface display of CD33FLbut, instead, expressed increased levels of the CD33∆E2transcript. CD33∆E2ML-1 cells were completely resistant to GO and AMG 330, whereas wild-type ML-1 cells were highly sensitive to these two drugs. Consistent with the findings in ML-1 cells, delivery of CRISPR/Cas9 RNPs to human fetal liver CD34+ HSPCs resulted in the expected CD33exon 2 deletion and decreased CD33FLsurface expression, while it did not impact the distribution of colony-forming cellsand only minimally reduced colony-forming potential. In NSG mouse xenotransplantation experiments, we found these CD33∆E2human CD34+ HSPCs to have comparable engraftment and multilineage differentiation potential relative to HSPCs expressing CD33FL(Fig. 1A). As expected, CD33FLexpression was substantially reduced in peripheral blood monocytes from CD33∆E2 HSPC-engrafted animals (Fig. 1B). In addition, robust engraftment of CD33∆E2 -engineered HSPCs in bone marrow was demonstrated by over 50% biallelic CD33exon 2 deletion at time of necropsy from colony-forming cells analysis (Fig. 1C). To determine if in vivodifferentiated myeloid CD33∆E2cells derived from infused human CD34+ HSPCs are indeed resistant to CD33-directed therapies, we treated mice with GO intravenously. GO administration reduced the number of circulating CD33FLCD14+ myeloid cells while leaving the number of CD33∆E2cells unaffected.
Conclusion:These findings support a novel strategy in which CD33∆E2-engineeredHSPCs are used to widen the therapeutic window of CD33-directed immunotherapies. Such cells could be envisioned for people at risk for the need of CD33-immunotherapy, e.g. AML patients in remission (where CD33-immunotherapy could be used to prevent/treat relapse) or, pre-emptively, for people with genetic AML-predisposition syndromes (such that CD33-targeted immunotherapy could be given safely if AML occurred). With this potential, further development of CD33∆E2-engineered HSPCs toward clinical application is warranted.
Walter:Amphivena Therapeutics, Inc: Consultancy, Other: Clinical Trial Support, Research Funding; Aptevo Therapeutics, Inc: Consultancy, Other: Clinical Trial Support, Research Funding; Covagen AG: Consultancy, Other: Clinical Trial Support, Research Funding; Actinium Pharmaceuticals, Inc: Other: Clinical Trial support , Research Funding; Amgen Inc: Other: Clinical Trial Support, Research Funding; Boehringer Ingelheim Pharma GmbH & Co. KG: Consultancy; Pfizer, Inc: Consultancy; Seattle Genetics, Inc: Consultancy, Other: Clinical Trial Support, Research Funding. Kiem:Magenta: Consultancy; Homology Medicine: Consultancy; Rocket Pharmaceuticals: Consultancy.

